
基本法规介绍
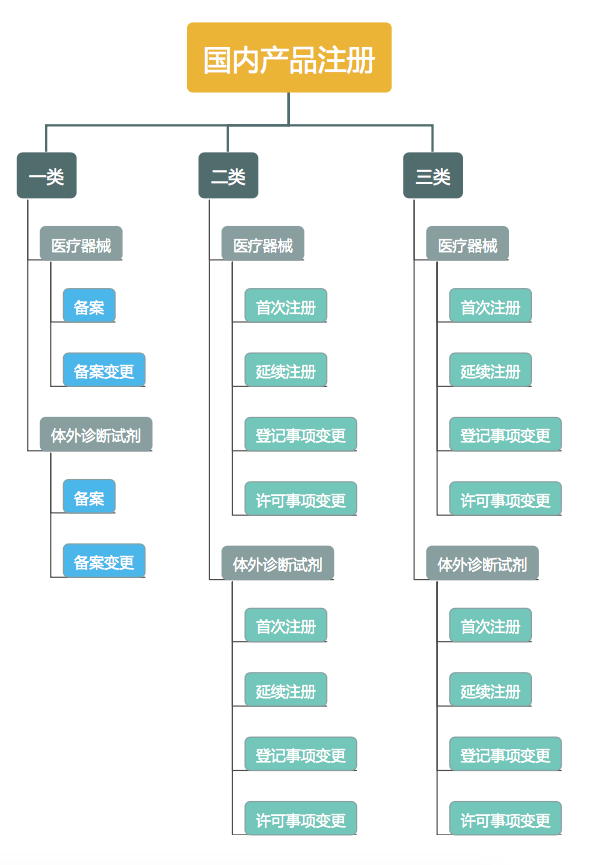
国家对医疗器械按照风险程度实行分类管理,分为一类、二类、三类:
- 第一类是风险程度低,实行常规管理可以保证其安全、有效的医疗器械。
- 第二类是具有中度风险,需要严格控制管理以保证其安全、有效的医疗器械。
- 第三类是具有较高风险,需要采取特别措施严格控制管理以保证其安全、有效的医疗器械。
第一类医疗器械实行备案管理。第二类、第三类医疗器械实行注册管理。
主要法规:
1、《医疗器械监督管理条例》(国务院令第680号)2017年5月4日发布
2、《医疗器械注册管理办法》(国家食品药品监督管理总局令第4号)2014年07月30日 发布
3、《体外诊断试剂注册管理办法》(国家食品药品监督管理总局令第5号)2014年07月30日 发布
服务项目介绍
首次注册
拟申报二、三类产品第一次在中国食品药品监督管理局(NMPA)办理注册工作。签发证书有效期五年。
延续注册
拟申报产品证书有效期届满前6个月应提出申请的延续证书有效期的工作。每次申请延续五年有效期。
注册变更
已注册的第二类、第三类医疗器械,医疗器械注册证及其附件载明的内容发生变化,注册人应当向原注册部门申请注册变更。注册变更分为登记事项变更、许可事项变更。
登记事项变更
注册人名称和住所、代理人名称和住所发生变化的,注册人应当向原注册部门申请登记事项变更;境内医疗器械生产地址变更的,注册人应当在相应的生产许可变更后办理注册登记事项变更。
许可事项变更
产品名称、型号、规格、结构及组成、适用范围、产品技术要求、进口医疗器械生产地址等发生变化的,注册人应当向原注册部门申请许可事项变更。
备案
第一类医疗器械生产前,应当办理产品备案。无有效期限制。
备案变更
第一类医疗器械经过备案的备案信息表中内容、技术要求内容发生变化的,备案人向原备案部门提出变更备案信息。
服务流程
简洁而完善的服务流程,助您迅速开启医疗器械合规之旅
您发起服务请求
电话联系400-88-45670即可。也可免费注册一个帐号,方便后面业务对接。
我们与您对接
我们接收到您的服务请求后,第一时间安排对口的专家及其工作小组与您对接联系。
全程服务跟踪
合作意向确定之后,服务小组启动一系列工作,全程跟踪整个项目直至完成。
关于「国内产品注册」的更多信息
合规常见问题
一般也包括中成药、中药材、保健品及医疗器材。
您需要提供审核人员的所有差旅费用。
根据您填写的仓库数目、现场数等基本信息,客服人员会尽快为您提供报价。
根据审核员老师的排期情况,一般2个月左右可安排审核。
在您关闭所有不符合项后,4到6周即可获证。
国家食品药品监督管理局对于风险等级的认定情况如下:
第一类是风险程度低,实行常规管理可以保证其安全、有效的医疗器械。故一类产品医疗器械实行产品备案管理。
第二类是具有中度风险,需要严格控制管理以保证其安全、有效的医疗器械。
第三类是具有较高风险,需要采取特别措施严格控制管理以保证其安全、有效的医疗器械。故第二类、第三类医疗器械实行产品注册管理。
第一类医疗器械产品备案和申请第二类、第三类医疗器械产品注册,应当提交下列资料:
1.产品风险分析资料;
2.产品技术要求;
3.产品检验报告;
4.临床评价资料;
5.产品说明书及标签样稿;
6.与产品研制、生产有关的质量管理体系文件;
7.证明产品安全、有效所需的其他资料。
医疗器械注册申请人、备案人应当对所提交资料的真实性负责。
除了西药,GDP也可以包括其他产品吗?
一般也包括中成药、中药材、保健品及医疗器材。
我们的合规专家库
-
王博维 - 医疗器械合规领域权威专家,擅长所有业务
感谢您联系奥斯曼!
我们的团队已收到您的消息,并将尽快回复您。

400-88-45670
请您拨打上面的电话号码